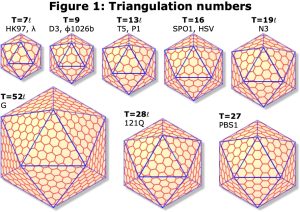
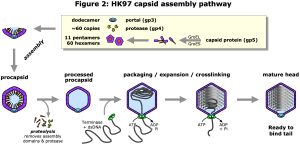
We use cryo-electron microscopy (cryoEM) to study the structure and function of protein complexes in their native, assembled and functional states. Advances in cryoEM technology and software analysis allow us to determine structures to atomic resolution (<1.8 Å) in ideal conditions, or “side-chain” resolution (<3.5Å) more generally where atomic models can be derived with confidence. A major focus is viruses, including human herpesviruses and the ubiquitous double-stranded-DNA (dsDNA) tailed bacteriophages. These two classes of virus have similar protein folds and assembly pathways indicating a deep evolutionary connection. Both assemble precursor protein shells, called procapsids, from many copies of a major capsid protein, a protein scaffold, and in some cases minor capsid proteins, all organized with icosahedral symmetry. The major capsid proteins organize as pentamers at the capsid vertices and hexamers elsewhere, with the number of hexamers varying according to capsid size (Figure 1). The scaffold is essential for this initial assembly, but after completion of the procapsid, a viral protease cleaves the scaffold leaving the procapsid ready to bind a terminase complex that packages the viral dsDNA into the capsid and triggers expansion to the mature capsid form (Figure 2). For herpesviruses, the DNA-filled capsid is assembled in the nucleus before budding out for subsequent maturation and egress as an enveloped virion. Tailed bacteriophages instead bind a separately assembled tail complex to form the infectious particle that is exported or lysed from the host bacterium.
Bacteriophage HK97 capsid structure
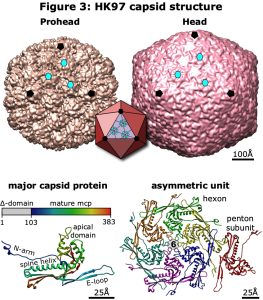
Since our initial genetic and cryoEM studies in the 1990’s, the phage HK97 has become a model for studying virus capsid structure and assembly. Xray crystallography and high-resolution cryoEM have resolved many aspects of the prohead and mature head structures, including the capsid geometry of T=7, the major capsid protein (mcp) fold, the expansion transition, and the network of covalent crosslinking between mcps in the mature capsid (Figure 3). The HK97 mcp is now recognized as common to all dsDNA phages and archaeal viruses, the eukaryotic herpesviruses, and bacterial compartments such as encapsulins, indicating a deep evolutionary connection between these different particles from all Domains of life. However, most structural studies of particles with the HK97 fold have exploited the capsid’s icosahedral symmetry to resolve density maps of sufficient quality for atomic modeling, but at the cost of masking the unique portal vertex. The portal is a 12-member ring occupying one of the icosahedral vertices and has critical functions in the virus lifecycle starting with nucleation of procapsid assembly. After completion of assembly, the scaffold is proteolysed and the terminase-DNA complex binds to the portal forming a pump that drives the dsDNA into the capsid to concentrations ~500 mg/mL powered by ATP hydrolysis. During this packaging process, the procapsid expands into the mature Head form, doubling the internal volume and initiating covalent crosslinking between mcps. When DNA packaging and capsid expansion are complete, the terminase disengages and the portal is closed to retain the encapsidated DNA. In phages, a tail structure binds to the portal, and during infection the portal is opened to release the viral DNA into a new host cell, or nucleus for herpesviruses.
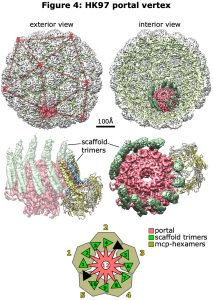
With recent advances in microscope and camera technology as well as analysis software, the portal vertex can be distinguished from the pentamer vertices allowing density maps of the portal vertex to be developed and modeled. This is a structurally curious feature of the capsid, where the icosahedral 5-fold symmetry is broken by the 12-fold portal ring, as well as being a functionally rich region involved in assembly, DNA packaging and retention, binding of the tail complex, and release of the DNA. These aspects are now under direct study, and our recent contribution has been to visualize the portal vertex in the initial procapsid form of bacteriophage HK97 before cleavage of the scaffold. HK97 is attractive for this work for several reasons – it’s host is E. coli and so easy to grow; the major capsid protein fold is known from Xray crystallography, the first of this large family to be solved; and the scaffold function is encoded in a 100-residue N-terminal extension of the major capsid protein, so it is present in a 1:1 ratio with the major capsid protein unlike other phages (and herpesviruses) that encode the scaffold on a separate gene. From cryoEM images of Prohead 1 (including scaffold) oriented by using icosahedral symmetry, the 12 vertices were compared to identify the unique portal vertex in each of 10,000s of particles. These were then mutually aligned to generate a density map revealing the organization of the portal vertex in this Prohead form (Figure 4). Fortunately, the well-resolved density indicated that all the particles adopted a single organization – not a given – and that the 5-12 symmetry mismatch was accommodated by the scaffold domains, as follows:
- the portal is surrounded by five hexamers of the major capsid protein
- the six scaffold domains in each hexamer form two trimeric coiled-coil towers
- thus, the five hexamers contribute 10 towers of scaffold that bind identically to the 12-fold portal
- the towers bind as pairs in a 4+6 arrangement, with two gaps where binding sites on the portal are unoccupied
- a short flexible linker between the coiled-coil domain and the C-terminal-most part of the scaffold domain allows for variations in positioning the towers identically against the portal subunits.
The 5-12-fold symmetry mismatch is more completely described as a progression of symmetries from 5-10-12-fold, and if the assembly is nucleated by the 12-subunit portal, then assembly starts with the 12-10 portal-scaffold accommodation to establish the overall 5-fold icosahedral vertex including five hexameric capsomers onto which the rest of the capsid assembles. We are following the maturation of this model by examining the procapsid after the scaffold has been proteolysed, and again after DNA packaging and capsid expansion. In addition, having refined our methods on the smaller and simpler HK97 capsid, we are now looking at phages with the HK97 fold and portal-scaffold-major capsid protein gene structure, but which make larger capsids.
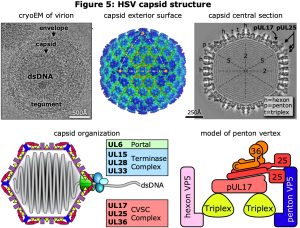
Herpesvirus capsid structure
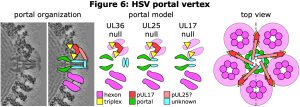
As for phage HK97, we have long collaborated with Fred Homa (also at U Pitt) to study the structure of herpesvirus capsids, particularly those of herpesvirus simplex type 1 (HSV1) and pseudorabies virus (PRV). Although the herpesvirus mcp is based on the HK97 fold, the HSV1 mcp, pUL19 (VP5), is much longer (1384 vs 384 amino acids) due to the insertion into the HK97 fold of a massive loop that forms chimney-like turrets extending from the capsid’s exterior surface. A number of additional minor capsid proteins include the triplex asymmetric trimer of pUL38 (VP19C) and pUL18 (VP23), the small hexon capping protein VP26, and the capsid vertex specific component (CVSC) of pUL17, pUL25 and pUL36 (Figure 5). The herpesvirus capsids are all larger than HK97, with T-number of 16 vs 7, but similarly include a portal at a unique vertex. We have applied our methods for selecting and resolving the portal vertex to compare capsids from virions with those of mutants lacking one of the three subunits of the essential CVSC molecule that binds around the penton vertices as well as the portal vertex (Figure 6). We found that lack of the UL17 protein prevents terminase binding and no DNA is packaged, while loss of UL25 allows DNA packaging to complete but the DNA is not retained inside the capsid. In contrast, the loss of UL36 produces a fully packaged capsid but it is unable to leave the nucleus for further maturation. Although we have insufficient images of capsid to dispense with 5-fold symmetry around the portal axis, and so have not yet resolved the portal correctly, we note that the portal is displaced by 30 Ångstroms radially after DNA is packaged (UL17-null vs virion), and that it returns to its starting position if the packaged DNA is released (UL25-null). Additional density around the portal vertex has not yet been visualized at sufficient detail for confident assignment, and this is a work in progress.
Cryo-electron microscopy (cryoEM)
CryoEM is a method of imaging protein complexes in their native or near-native state without chemical fixation or dehydration, thus preserving structure. To achieve this, very thin layers of aqueous sample are suspended over holes in a graphitized carbon support film and vitrified (ie, frozen without forming crystals) by plunging rapidly into liquid ethane. Frozen grids are transferred into the cryo-electron microscope at liquid nitrogen temperature to maintain the vitrified state and are imaged with low doses of electron radiation to avoid damage. Images represent projected density, and three-dimensional representations of the original structure can be obtained by back-projection or equivalent methods.
Single-particle methods (somewhat mis-named) involve collecting thousands of micrographs with thousands to millions of images of individual particles that are assumed to be in a uniform state, or at most in several distinct states. Particle images that resemble each other, save for translation and in-plane rotation, are collected into groups and these groups are combined in a three-dimensional sense to resolve a preliminary structure. The resolution and quality of the structure is improved by refinement of the orientations assigned to each particle, and microscope and camera distortions are also accounted for. This method has yielded the highest resolution structures to date, including apoferritin (a test sample) to 1.2 Ångstroms. Particular successes include icosahedral virus particles, which are difficult for other methods due to their size, membrane proteins that are difficult to crystallize, and amyloid fibrils.
Tomography is used to collect a tilt series of images of the same object, typically a non-uniform sample such as a bacterium or organelle, and is otherwise similar to the single particle methods in using back-projection to calculate three-dimensional models from the tilt series. Resolution is limited by the large electron dose used in collecting data, but developments such as sub-tomogram averaging continue to advance the reach of cryo-electron tomography. When samples may be too thick for electrons to penetrate, thin sections of interest, called lamellae, may be milled from the frozen sample using a Focused Ion Beam mill (FIB-mill), which is a scanning electron microscope equipped with a second beam of ions that perform the milling.